
We have now collected blood from the bird in the field, purified a DNA sample from that blood, and amplified billions of copies of a target locus from that DNA. The next step, the second step heavy in biochemistry, is reading the sequence of that DNA strand.
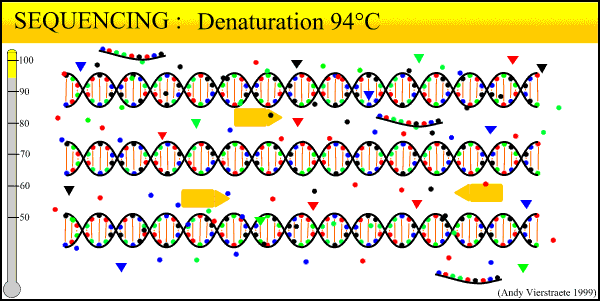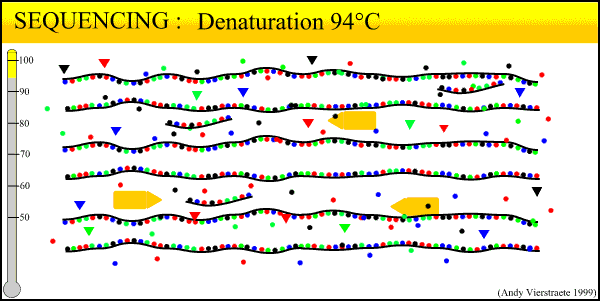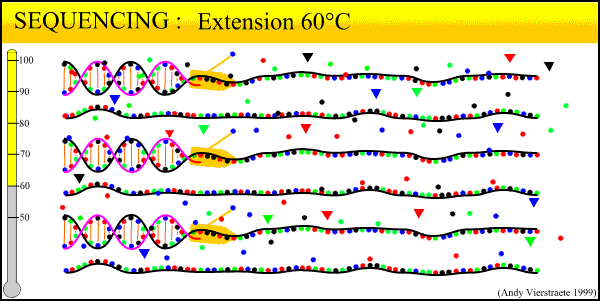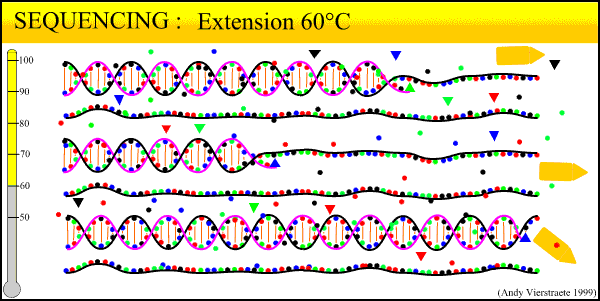
The sequencing method now widely used is known as dye-terminator sequencing, and is a derivation of the original Sanger sequencing. The process begins with a reaction that is mechanistically much the same as PCR, with a series of three-step temperature cycles to create and elongate new DNA strands. This time, ddNTPs (dideoxynucleotide triphosphate) are added to the mixture of dNTPs (deoxynucleotide triphosphate). These nucleotides are identical to those normally used in DNA elongation, except that they lack the -OH hydroxyl group used by the polymerase to add the next nucleotide in the sequence. Thus, when added they terminate the sequence. These ddNTPs have a fluorescent dye attached, with a different color of fluorescence for each base (A, T, C, G), which will be used later to actually read the sequence. The ddNTPs are added in much smaller concentrations than the dNTPs, so the cycling produces a variety of lengths of sequence, depending on when a ddNTP happens to be added instead of a dNTP. Finally, another difference from PCR is the primer mixture used. In the sequencing reaction, only one primer is added to the mixture, instead of both the forward and reverse primers being added. The reason for this will become clear later.
An animated image of the sequencing reaction can be found here.
What results from this process is a mixture of fragments that start with either the forward or reverse primer, proceed in the direction of the primer for a random length, and terminate in a dye-labeled ddNTP. The number of new fragments produced in the sequencing reaction is not nearly as much as in regular PCR, even when run for 30 cycles. This is because only one primer is used at a time during the reaction. The reaction can only proceed in one direction, and the number of copies produced increases linearly rather than exponentially. Thus, the exponential amplification of the locus in PCR beforehand is needed to generate enough copies for the sequencer to read.
An animated image of the sequencing reaction can be found here.
{kind=link}
What results from this process is a mixture of fragments that start with either the forward or reverse primer, proceed in the direction of the primer for a random length, and terminate in a dye-labeled ddNTP. The number of new fragments produced in the sequencing reaction is not nearly as much as in regular PCR, even when run for 30 cycles. This is because only one primer is used at a time during the reaction. The reaction can only proceed in one direction, and the number of copies produced increases linearly rather than exponentially. Thus, the exponential amplification of the locus in PCR beforehand is needed to generate enough copies for the sequencer to read.
The sequencer

Above is our lab's sequencer, a small unit that can only run sixteen samples at a time. We generally run our sequencing at another lab on campus, which has sequencers that can run whole 96-well plates at a time, taking about 3 hours. The samples from the sequencing reaction are mixed into a kind of gel and are read by the sequencer in a process similar to gel electrophoresis. Remember the sample produced in the sequencing reaction is a mixture of fragments of varying lengths. Given the number of copies involved, fragments exist for every length out to several hundred base pairs. When an electrical current is applied, the negative charge of DNA acts to pull it towards the positive node.
The sequencer pulls the DNA fragments through a gel in a tiny capillary tube. The smaller fragments travel faster and farther through the gel. This organizes the fragment mixture pulled through the capillary by length, smallest first. At the end of the capillaries, there is a small sensor that reads the wavelength of fluorescence of the gel. By the time the fragments go past the sensor, they have been pulled into discrete clusters and the sensor reads a peak of color. Thus the first fragment (label on base pair 1) is read, then the second fragment (label on base pair 2), and so on until out of fragments.
The sequencer pulls the DNA fragments through a gel in a tiny capillary tube. The smaller fragments travel faster and farther through the gel. This organizes the fragment mixture pulled through the capillary by length, smallest first. At the end of the capillaries, there is a small sensor that reads the wavelength of fluorescence of the gel. By the time the fragments go past the sensor, they have been pulled into discrete clusters and the sensor reads a peak of color. Thus the first fragment (label on base pair 1) is read, then the second fragment (label on base pair 2), and so on until out of fragments.
Capillaries for 16 samples

The sequencer's all-important sensor

What this process boils down to: reading DNA sequence is as simple as labeling each base pair with a particular color, and drawing the strand past a sensor that reads the color. The sequencer outputs the data to a computer. Using programs such as Sequencher, we then 'clean up the data' for analysis. The output looks as follows:
 (Crop from Source)
(Crop from Source){kind=link}
We can view both the raw data, the colored peaks, and the program's preliminary base 'calls' (its best guess for each peak). 'Cleaning' the data consists of scrolling through the sequence, looking for ambiguous areas caused by dye blobs or other malfunctions. In the example above, the top row of peaks is extremely messy and difficult to call. The bottom row is much cleaner, with well-defined peaks, although there is a very low level of 'noise', the low amounts of blob along the bottom. Too much noise can make the sequence unreadable. Also, true double peaks can occur in heterozygotes, where the two copies of the locus (remember each gene has two copies) differ at a base pair or even a long deletion or insertion. Rerunning the sample will fix most errors; true double peaks can be resolved by cloning (where one copy of the locus is cloned into a bacterium and then sequenced) or by running a program such as PHASE to estimate the sequence of the differing alleles.
Remember I mentioned earlier that a single primer is run in the sequencing reaction, and that both a forward and reverse primer is run for each sample (thus, two sequencing reactions per sample). Besides being necessary for the sequencing reaction to work, this solves two problems in the clean-up phase. The first is length: sequencing reactions generally don't produce fragments the length of the whole locus, so by having two sequences starting the beginning and the end we can reconstruct the whole locus. The second is noise: as the sequencing fragments get longer, the number of copies produced is fewer and thus the quality of the peak read by the sequencer degrades. Having overlapping forward and reverse fragments allows us to call peaks with greater certainty, by having two strands of peaks to call from.
When all of the data is cleaned up, with the ambiguous peaks resolved and any heterozygous alleles identified and sequenced, we now have our data set! We are ready for analysis.
Part 5: Data Analysis and Results
Remember I mentioned earlier that a single primer is run in the sequencing reaction, and that both a forward and reverse primer is run for each sample (thus, two sequencing reactions per sample). Besides being necessary for the sequencing reaction to work, this solves two problems in the clean-up phase. The first is length: sequencing reactions generally don't produce fragments the length of the whole locus, so by having two sequences starting the beginning and the end we can reconstruct the whole locus. The second is noise: as the sequencing fragments get longer, the number of copies produced is fewer and thus the quality of the peak read by the sequencer degrades. Having overlapping forward and reverse fragments allows us to call peaks with greater certainty, by having two strands of peaks to call from.
When all of the data is cleaned up, with the ambiguous peaks resolved and any heterozygous alleles identified and sequenced, we now have our data set! We are ready for analysis.
Part 5: Data Analysis and Results
No comments:
Post a Comment